A Complete Guide to Single-Cell ATAC Sequencing Data Analysis
📋 Overview • 📁 File Preparation • 📊 Reference Data • 🚀 Main Pipeline • 📊 Results Interpretation
This document provides a detailed guide for analyzing single-cell ATAC sequencing data using dnbc4tools.
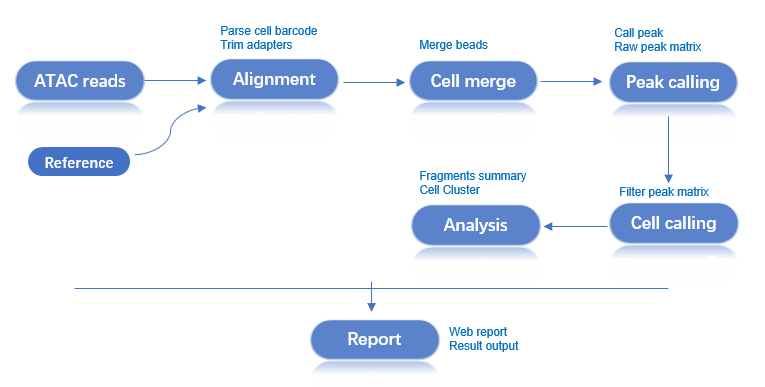
Workflow: Raw Data → Quality Control → Alignment → Bead Merging → Peak Calling → Cell Identification → Dimensionality Reduction & Clustering → Analysis Report
The analysis requires FASTQ files:
| File Type | Description |
|---|---|
| ATAC Library | Sequencing data containing cell barcodes and chromatin accessibility information. |
| File Type | Format | Description |
|---|---|---|
| Genome File | FASTA | Contains the complete genome sequence of a species, including chromosomes, mitochondria, and other genetic information, typically the primary assembly. This file provides the foundation for genome analysis and alignment. |
| Annotation File | GTF | Contains detailed information about genes, transcripts, exons, and other functional regions in the genome. This file identifies the location, type, and related attributes of genes. |
GTF File Requirements:
For details on GTF file filtering, please refer to the scRNA analysis pipeline.
Before running the dnbc4tools atac run analysis, a reference database must be built. This step requires an annotation file (GTF) and a reference genome (FASTA) to create index files for read alignment and statistical analysis.
$dnbc4tools atac mkref \
--fasta genome.fa \
--ingtf genes.gtf \
--species Mus_musculus
Output:
Upon successful execution, a reference database directory will be created at the specified location with the following structure:
/opt/database/Mus_musculus
├── fasta
│ ├── genome.fa
│ ├── genome.fa.fai
│ ├── genome.index
│ └── genome.index.log
├── genes
│ └── genes.gtf
├── ref.json
└── regions
├── chrom.sizes
├── promoter.bed
└── tss.bed
The ref.json file records the main information of the database:
{
"species": "Mus_musculus",
"input_fasta_files": [
"genome.fa"
],
"input_gtf_files": [
"genes.gtf"
],
"genome": "/opt/database/Mus_musculus/fasta/genome.fa",
"index": "/opt/database/Mus_musculus/fasta/genome.index",
"gtf": "/opt/database/Mus_musculus/genes/genes.gtf",
"chrmt": "chrM",
"chloroplast": "None",
"chromeSize": "/opt/database/Mus_musculus/regions/chrom.sizes",
"tss": "/opt/database/Mus_musculus/regions/tss.bed",
"promoter": "/opt/database/Mus_musculus/regions/promoter.bed",
"version": "dnbc4tools 3.0beta",
"blacklist": "None",
"genomesize": "mm"
}
The following information will be printed during runtime:
Creating new reference folder at /opt/database/Mus_musculus
...done
Writing genome FASTA file into reference folder...
...done
Indexing genome FASTA file...
...done
Writing genes GTF file into reference folder...
...done
Extracting TSS and promoter regions from GTF file...
...done
Generating Chromap genome index...
...done
Writing reference JSON file...
...done
Analysis Complete
To simplify the analysis of multiple samples, you can use a configuration file to generate a shell script for each sample.
$dnbc4tools atac multi \
--list sample.tsv \
--genomeDir /opt/database/Mus_musculus \
--threads 10
The sample.tsv file is tab-separated (\t) and contains two columns:
| Column | Content |
|---|---|
| 1 | Sample Name |
| 2 | Library Sequencing Data |
sample1 /data/sample1_R1.fq.gz;/data/sample1_R2.fq.gz
sample2 /data/sample2_R1.fq.gz;/data/sample2_R2.fq.gz
sample3 /data/sample3_1_R1.fq.gz,/data/sample3_2_R1.fq.gz;/data/sample3_1_R2.fq.gz,/data/sample3_2_R2.fq.gz
After execution, a shell script is generated for each sample:
sample1.sh
sample2.sh
sample3.sh
Example content of sample1.sh:
$cat sample1.sh
/opt/software/dnbc4tools3.0Beta/dnbc4tools atac run --name sample1 --fastq1 /data/sample1_R1.fq.gz --fastq2 /data/sample1_R2.fq.gz --genomeDir /opt/database/Mus_musculus --threads 10
You can then execute these scripts to run the main analysis.
The ATAC main analysis pipeline processes single-cell ATAC library data from a single sample. It filters and aligns reads to generate a fragments file for all beads. Beads are then merged, and peak calling is performed. Cell identification is done using the fragment information within the peak regions. This is followed by cell filtering, dimensionality reduction, and clustering. Finally, the results from all steps are integrated to generate an HTML report and other output files.
Example script for generating an expression matrix for a single sample:
$dnbc4tools atac run \
--name sample \
--fastq1 /sample/data/test1_R1.fastq.gz,/sample/data/test2_R1.fastq.gz \
--fastq2 /sample/data/test1_R2.fastq.gz,/sample/data/test2_R2.fastq.gz \
--genomeDir /opt/database/Mus_musculus \
--threads 10
After auto-detecting the reagent version and dark reaction, the software begins the analysis. Here is an example:
2025-06-03 16:24:27 Performing ATAC data processing
Chemistry(darkreaction) determined in fastqR1: darkreaction
Chemistry(darkreaction) determined in fastqR2: darkreaction
2025-06-03 16:24:30 Performing quality control and alignment on raw data...
...done
2025-06-03 16:36:25 Computing bead similarity and merging beads within droplets...
...done
2025-06-03 16:38:21 Processing fragments for peak calling...
...done
2025-06-03 16:40:06 Generating raw peaks matrix...
...done
2025-06-03 16:47:30 Generating filtered peaks matrix...
...done
2025-06-03 16:50:52 Conducting dimensionality reduction and clustering...
...done
2025-06-03 16:54:44 Statistical analysis and report generation for results...
...done
Analysis Finished Elapsed Time: 0:30:43
A successful run will end with Analysis Finished.
Upon completion, outs (outputs) and logs directories will be generated.
.
├── *_scATAC_report.html
├── filter_peak_matrix/
│ ├── barcodes.tsv.gz
│ ├── matrix.mtx.gz
│ └── peaks.bed.gz
├── fragments.tsv.gz
├── fragments.tsv.gz.tbi
├── metrics_summary.xls
├── raw_peak_matrix/
│ ├── barcodes.tsv.gz
│ ├── matrix.mtx.gz
│ └── peaks.bed.gz
└── singlecell.csv
Related Documentation:
Content to be added