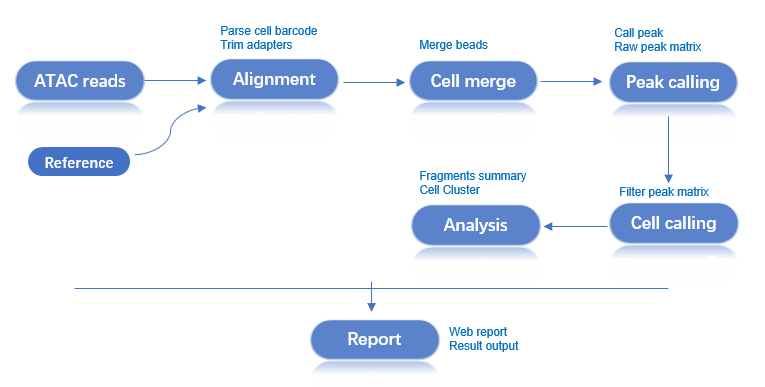
本文档详细介绍了使用 dnbc4tools 进行单细胞 ATAC 测序数据分析的完整流程。
工作流程:原始数据 → 质量控制 → 比对 → 磁珠合并 → Peak调用 → 细胞识别 → 降维聚类 → 分析报告
分析需要FASTQ文件:
| 文件类型 | 说明 |
|---|---|
| ATAC文库 | 包含cell barcode和染色质开放区域信息的测序数据 |
| 文件类型 | 格式 | 说明 |
|---|---|---|
| 基因组文件 | FASTA | 包含特定物种的完整基因组序列,包括染色体、线粒体及其他遗传信息,通常为主装配版本。这些文件为基因组分析和比对提供基础数据。 |
| 注释文件 | GTF | 包含基因组中基因、转录本、外显子及其他功能区域的详细信息。该文件标识基因的位置、类型及其相关属性。 |
GTF文件要求:
有关GTF文件过滤的详细信息,请参考scRNA分析流程。
在运行dnbc4tools atac run分析之前,我们需要优先构建参考数据库。此步骤需要注释文件(GTF)和参考基因组(FASTA)来构建索引文件,用于测序reads的比对和统计分析。
$dnbc4tools atac mkref \
--fasta genome.fa \
--ingtf genes.gtf \
--species Mus_musculus
输出结果:
成功运行后,将在指定位置创建参考数据库目录,包含以下文件结构:
/opt/database/Mus_musculus
├── fasta
│ ├── genome.fa
│ ├── genome.fa.fai
│ ├── genome.index
│ └── genome.index.log
├── genes
│ └── genes.gtf
├── ref.json
└── regions
├── chrom.sizes
├── promoter.bed
└── tss.bed
其中ref.json文件中记录数据库的主要信息:
{
"species": "Mus_musculus",
"input_fasta_files": [
"genome.fa"
],
"input_gtf_files": [
"genes.gtf"
],
"genome": "/opt/database/Mus_musculus/fasta/genome.fa",
"index": "/opt/database/Mus_musculus/fasta/genome.index",
"gtf": "/opt/database/Mus_musculus/genes/genes.gtf",
"chrmt": "chrM",
"chloroplast": "None",
"chromeSize": "/opt/database/Mus_musculus/regions/chrom.sizes",
"tss": "/opt/database/Mus_musculus/regions/tss.bed",
"promoter": "/opt/database/Mus_musculus/regions/promoter.bed",
"version": "dnbc4tools 3.0beta",
"blacklist": "None",
"genomesize": "mm"
}
运行时打印信息,以下是一个示例:
Creating new reference folder at /opt/database/Mus_musculus
...done
Writing genome FASTA file into reference folder...
...done
Indexing genome FASTA file...
...done
Writing genes GTF file into reference folder...
...done
Extracting TSS and promoter regions from GTF file...
...done
Generating Chromap genome index...
...done
Writing reference JSON file...
...done
Analysis Complete
为了简化每个样本单独生成主分析流程,可以使用配置文件来生成一个包含多个样本的主流程 shell 脚本。以下是一个示例步骤或脚本模板:
$dnbc4tools atac multi \
--list sample.tsv \
--genomeDir /opt/database/Mus_musculus \
--threads 10
其中 sample.tsv 文件使用制表符 (\t) 分隔,包含两列:
| 列 | 内容 |
|---|---|
| 1 | 样本名称 |
| 2 | 文库测序数据 |
sample1 /data/sample1_R1.fq.gz;/data/sample1_R2.fq.gz
sample2 /data/sample2_R1.fq.gz;/data/sample2_R2.fq.gz
sample3 /data/sample3_1_R1.fq.gz,/data/sample3_2_R1.fq.gz;/data/sample3_1_R2.fq.gz,/data/sample3_2_R2.fq.gz
运行完成后输出:
sample1.sh
sample2.sh
sample3.sh
其中文件 sample1.sh 如下:
$cat sample1.sh
/opt/software/dnbc4tools3.0Beta/dnbc4tools atac run --name sample1 --fastq1 /data/sample1_R1.fq.gz --fastq2 /data/sample1_R2.fq.gz --genomeDir /opt/database/Mus_musculus --threads 10
执行第四步进行主流程分析。
ATAC 主分析流程使用单个样本单细胞 ATAC 文库测序数据,经过过滤和比对生成所有磁珠的 fragments 文件。合并磁珠并执行 peak 调用分析,利用 peaks 区域的片段信息进行细胞识别。随后进行细胞过滤、降维和聚类,最终整合各步骤结果生成 HTML 网页报告并输出分析结果。
为单个样本生成表达矩阵,以下是一个示例步骤或脚本模板:
$dnbc4tools atac run \
--name sample \
--fastq1 /sample/data/test1_R1.fastq.gz,/sample/data/test2_R1.fastq.gz \
--fastq2 /sample/data/test1_R2.fastq.gz,/sample/data/test2_R2.fastq.gz \
--genomeDir /opt/database/Mus_musculus \
--threads 10
在对试剂版本和暗反应自动检测后,软件开始运行分析,以下是一个示例:
2025-06-03 16:24:27 Performing ATAC data processing
Chemistry(darkreaction) determined in fastqR1: darkreaction
Chemistry(darkreaction) determined in fastqR2: darkreaction
2025-06-03 16:24:30 Performing quality control and alignment on raw data...
...done
2025-06-03 16:36:25 Computing bead similarity and merging beads within droplets...
...done
2025-06-03 16:38:21 Processing fragments for peak calling...
...done
2025-06-03 16:40:06 Generating raw peaks matrix...
...done
2025-06-03 16:47:30 Generating filtered peaks matrix...
...done
2025-06-03 16:50:52 Conducting dimensionality reduction and clustering...
...done
2025-06-03 16:54:44 Statistical analysis and report generation for results...
...done
Analysis Finished Elapsed Time: 0:30:43
成功的运行会以 Analysis Finished 结束。
分析完成后,将生成结果输出目录outs,logs日志目录。
.
├── *_scATAC_report.html
├── filter_peak_matrix/
│ ├── barcodes.tsv.gz
│ ├── matrix.mtx.gz
│ └── peaks.bed.gz
├── fragments.tsv.gz
├── fragments.tsv.gz.tbi
├── metrics_summary.xls
├── raw_peak_matrix/
│ ├── barcodes.tsv.gz
│ ├── matrix.mtx.gz
│ └── peaks.bed.gz
└── singlecell.csv
相关文档:
内容待补充