A Complete Guide to Single-Cell RNA Sequencing Data Analysis
📋 Overview • 📁 File Preparation • 📊 Reference Data • 🚀 Main Pipeline • 📊 Results Interpretation
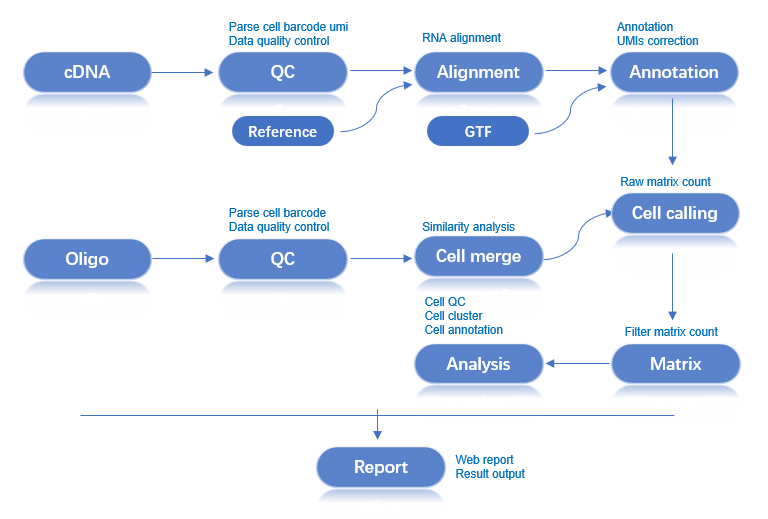
This document provides a complete guide on how to use dnbc4tools for single-cell RNA sequencing data analysis.
Workflow: Raw Data → Quality Control → Alignment → Cell Identification → Expression Matrix → Analysis Report
Two types of FASTQ files are required for the analysis:
| File Type | Description |
|---|---|
| cDNA Library | Sequencing data containing Cell Barcodes, UMIs, and transcriptome information. |
| Oligo Library | Contains Cell Barcode information for both large and small beads, and UMI information for small beads. |
| File Type | Format | Description |
|---|---|---|
| Genome File | FASTA | Contains the complete genome sequence of a species (usually the primary assembly), including chromosomes, mitochondria, and other genetic information. This file is fundamental for genome alignment and analysis. |
| Annotation File | GTF | Provides detailed information on genes, transcripts, exons, and other functional regions in the genome. This file specifies the location, type, and attributes of genes. |
GTF File Requirements:
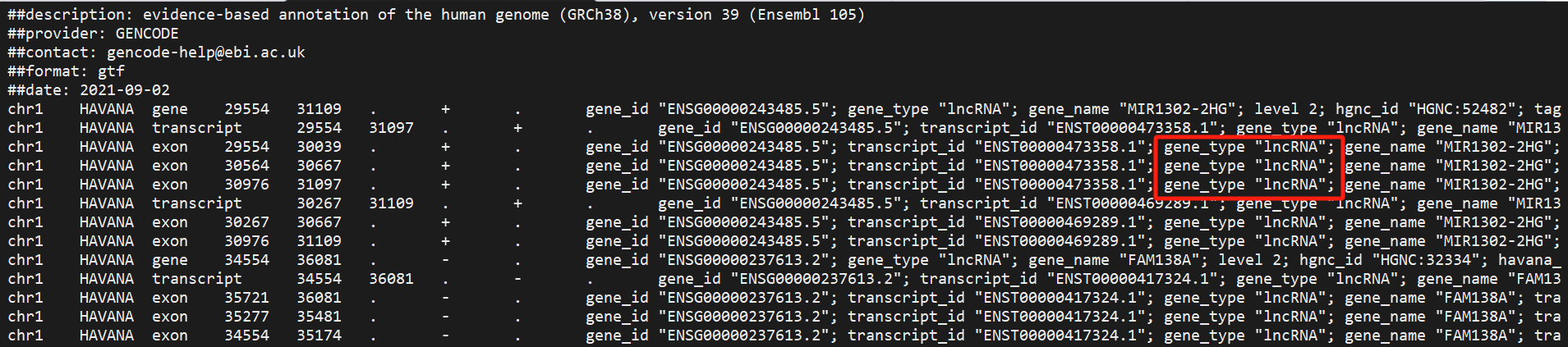
GTF files downloaded from sites like ENSEMBL and UCSC often contain many types of genes. Selecting specific gene types relevant to your research can reduce overlapping gene annotations and improve the uniqueness of alignments, as reads mapping non-uniquely to multiple genes are filtered out.
We provide three functions for GTF file processing:
| Function | Description |
|---|---|
| Gene Type Statistics | Count the number of various gene types in a GTF file. |
| Correct GTF File | Fill in missing information to ensure the GTF file meets analysis requirements. |
| Filter Gene Types | Filter specific gene types based on research needs. |
# Count gene type quantities
$dnbc4tools tools mkgtf \
--action stat \
--ingtf genes.gtf \
--output gtfstat.txt \
--type gene_biotype
Example Output:
$cat gtf_type.txt
Type Count
protein_coding 20006
lncRNA 17755
processed_pseudogene 10159
unprocessed_pseudogene 2605
misc_RNA 2221
snRNA 1910
miRNA 1879
TEC 1056
transcribed_unprocessed_pseudogene 950
snoRNA 943
transcribed_processed_pseudogene 503
rRNA_pseudogene 497
IG_V_pseudogene 187
transcribed_unitary_pseudogene 146
IG_V_gene 145
......
If a GTF file has incomplete content, the main analysis pipeline may fail due to incomplete annotations. This function can automatically fill in missing information in gene and transcript entries to ensure the pipeline runs smoothly.
# Correct GTF file
$dnbc4tools tools mkgtf \
--action check \
--ingtf genes.gtf \
--output corrected.gtf
The software cross-references gene_id with gene_name and transcript_id with transcript_name to fill in missing details and flags locations where multiple gene information might exist.
# Filter gene types
$dnbc4tools tools mkgtf \
--ingtf genes.gtf \
--output genes.filter.gtf \
--type gene_biotype
Default included gene types:
protein_coding
lncRNA/lincRNA
antisense
IG_V_gene
IG_LV_gene
IG_D_gene
IG_J_gene
IG_C_gene
IG_V_pseudogene
IG_J_pseudogene
IG_C_pseudogene
TR_V_gene
TR_D_gene
TR_J_gene
TR_C_gene
You can also use the include parameter to customize the gene types to be retained:
# Custom gene type filtering
$dnbc4tools tools mkgtf \
--ingtf genes.gtf \
--output genes.filter.gtf \
--type gene_biotype \
--include protein_coding,lncRNA,lincRNA,\
antisense,IG_V_gene,IG_LV_gene,IG_J_gene,\
IG_C_gene,IG_V_pseudogene,IG_J_pseudogene,\
IG_C_pseudogene,TR_V_gene,TR_D_gene,TR_J_gene,TR_C_gene
Before running the dnbc4tools rna run analysis, a reference database must be built. This step uses the annotation file (GTF) and reference genome (FASTA) to create an index for aligning and annotating the sequencing reads.
# Build reference database
$dnbc4tools rna mkref \
--fasta genome.fa \
--ingtf genes.gtf \
--species Homo_sapiens \
--threads 10
Output:
Upon successful execution, a reference database directory will be created at the specified location with the following structure:
/opt/database/Homo_sapiens
├── fasta
│ ├── genome.fa
│ └── genome.fa.fai
├── genes
│ └── genes.gtf
├── ref.json
└── star
├── chrLength.txt
├── chrNameLength.txt
├── chrName.txt
├── chrStart.txt
├── exonGeTrInfo.tab
├── exonInfo.tab
├── geneInfo.tab
├── Genome
├── genomeParameters.txt
├── mtgene.list
├── SA
├── SAindex
├── sjdbInfo.txt
├── sjdbList.fromGTF.out.tab
├── sjdbList.out.tab
└── transcriptInfo.tab
The ref.json file records the main information of the database.
{
"chrmt": "chrM",
"genome": "/opt/database/Homo_sapiens/fasta/genome.fa",
"genomeDir": "/opt/database/Homo_sapiens/star",
"gtf": "/opt/database/Homo_sapiens/genes/genes.gtf",
"input_fasta_files": [
"genome.fa"
],
"input_gtf_files": [
"genes.filter.gtf"
],
"mtgenes": "/opt/database/Homo_sapiens/star/mtgene.list",
"species": "Homo_sapiens",
"version": "dnbc4tools 3.0beta"
}
The following information will be printed during runtime:
Creating new reference folder at /opt/database/Homo_sapiens
...done
Writing genome FASTA file into reference folder...
...done
Indexing genome FASTA file...
...done
Writing genes GTF file into reference folder...
...done
Generating STAR genome index...
...done.
Writing Reference JSON file into reference folder...
...done
Analysis Complete
To simplify the analysis of multiple samples, you can use a configuration file to generate a shell script for each sample.
$dnbc4tools rna multi \
--list sample.tsv \
--genomeDir /opt/database/Homo_sapiens \
--threads 30
The sample.tsv file is tab-separated (\t) and contains three columns:
| Column | Content |
|---|---|
| 1 | Sample Name |
| 2 | cDNA Library Sequencing Data |
| 3 | Oligo Library Sequencing Data |
sample1 /data/cDNA1_R1.fq.gz;/data/cDNA1_R2.fq.gz /data/oligo1_R1.fq.gz,/data/oligo4_R1.fq.gz;/data/oligo1_R2.fq.gz,/data/oligo4_R2.fq.gz
sample2 /data/cDNA2_R1.fq.gz;/data/cDNA2_R2.fq.gz /data/oligo2_R1.fq.gz;/data/oligo2_R2.fq.gz
sample3 /data/cDNA3_R1.fq.gz;/data/cDNA3_R2.fq.gz /data/oligo3_R1.fq.gz;/data/oligo3_R2.fq.gz
After execution, a shell script will be generated for each sample:
sample1.sh
sample2.sh
sample3.sh
Example content of sample1.sh:
$cat sample1.sh
/opt/software/dnbc4tools3.0beta/dnbc4tools rna run --name sample1 --cDNAfastq1 /data/cDNA1_R1.fq.gz --cDNAfastq2 /data/cDNA1_R2.fq.gz --oligofastq1 /data/oligo1_R1.fq.gz,/data/oligo4_R1.fq.gz --oligofastq2 /data/oligo1_R2.fq.gz,/data/oligo4_R2.fq.gz --genomeDir /database/scRNA/Mus_musculus/mm10 --threads 30
You can then execute these scripts to run the main analysis.
The main RNA analysis pipeline processes cDNA and Oligo library data from a single sample. Key steps include:
Example script for generating an expression matrix for a single sample:
$dnbc4tools rna run \
--name sample \
--cDNAfastq1 /data/sample_cDNA_R1.fastq.gz \
--cDNAfastq2 /data/sample_cDNA_R2.fastq.gz \
--oligofastq1 /data/sample_oligo1_1.fq.gz,/data/sample_oligo2_1.fq.gz \
--oligofastq2 /data/sample_oligo1_2.fq.gz,/data/sample_oligo2_2.fq.gz \
--genomeDir /opt/database/Homo_sapiens \
--threads 30
After auto-detecting the reagent version and dark reaction, the software begins the analysis. Here is an example:
2025-06-04 16:29:35 Performing RNA data processing
Chemistry(darkreaction) determined in oligoR1: darkreaction
Chemistry(darkreaction) determined in oligoR2: darkreaction
Chemistry(darkreaction) determined in cDNAR1: darkreaction
2025-06-04 16:29:37 Processing oligo library filtering...
...done
2025-06-04 16:59:39 Processing cDNA library filtering...
...done
2025-06-04 17:50:10 Processing alignment and counting...
...done
2025-06-05 01:20:56 Calculating bead similarity, merging beads within the same droplet...
...done
2025-06-05 01:22:17 Generating raw gene expression matrix...
...done
2025-06-05 01:31:38 Generating cell-filtered gene expression matrix...
...done
2025-06-05 01:33:07 Calculating sequencing saturation metrics...
...done
2025-06-05 01:34:23 Generating position-sorted BAM file...
...done
2025-06-05 02:23:17 Performing dimensionality reduction and clustering analysis...
...done
2025-06-05 02:24:57 Generating analysis report and summary statistics...
...done
Analysis Finished
Elapsed Time: 9:56:09
When the message Analysis Finished appears, the analysis is successfully completed.
Upon completion, outs (outputs) and logs directories will be generated. The outs directory is structured as follows:
.
├── analysis/
│ ├── cluster.csv
│ ├── marker.csv
│ └── QC_Cluster.h5ad
├── anno_decon_sorted.bam
├── anno_decon_sorted.bam.bai
├── filter_feature.h5ad
├── filter_matrix/
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── metrics_summary.xls
├── raw_matrix/
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── *_scRNA_report.html
└── singlecell.csv
Related Documentation:
Content to be added